This is due to STK295900-induced accumulation of 4N DNA content with no PI-103 PI3K inhibitor significant change in mitotic index. In addition, STK295900-induced G2 arrest was confirmed by investigating the cell cycle regulatory proteins. Progression through the eukaryotic cell cycle is driven in part by a subfamily of Cdks whose BAY-60-7550 activities are modulated by forming bipartite complexes with different cyclins. Levels of cyclins oscillate throughout the cell cycle whereas Cdk protein levels remain stable. Therefore, the activity of Cdks is regulated by the presence of different cyclins. In mitotic cells, cyclin B1 level was relatively high whereas cyclin A was undetectable. In contrast, STK295900 showed similar effect as camptothecin and etoposide did on cyclin A and cyclin B1 accumulation without induction of Histone H3 phosphorylation at S10, which is crucial for chromosome condensation and cell-cycle progression during mitosis. STK295900 belongs to a class of symmetric bibenzimidazole group. Compounds containing benzimidazole ring have been used extensively for pharmacological purposes such as antimicrobial and pyridine-chemical-structure.gif) anticancer agents. Several asymmetric, head-to-tail bibenzimidazole derivatives, such as Hoechst 33258 and Hoechst 33342, exhibited antitumor activity by binding to minor groove of DNA at three consecutive A:T base pairs, leading to the inhibition of Top 1 activity. In addition, the symmetric bibenzimidazole derivatives, containing two groups of benzimidazole linked in head-to-head fashion, have been reported that they bind DNA minor groove with extending the binding site to four A:T base pairs and exhibit antitumor activity. However, there is no report on the mechanism of action for their antitumor activity. Here, we showed that STK295900 exerted its activity by interfering with Top 1 and Top 2 activities. In support of this notion, STK295900 was recently reported as a potent antistaphylococcal agent by targeting DNA gyrase. The results from DNA relaxation assay suggested that STK295900 stabilizes the DNA-Top 1 cleavable complex, a characteristic of Top poisons, but it also inhibited Top 2 catalytic activity. Generally, Top poisons causes DNA strand break and consequently triggers G2 arrest via activation of ATM/ATR signaling pathway. These kinases phosphorylate and activate Chk1 and Chk2, which in turn phosphorylate and inactivate Cdc25C phosphatase resulting in blocking the activation of Cdk1 and transition into mitosis. Zinc finger nucleases are artificial restriction enzymes that are comprised of custom-designed zinc finger proteins and a nuclease domain derived from the FokI endonuclease. Zinc finger proteins can be designed to bind to specific sequences of DNA, allowing ZFNs to induce double- or single-strand breaks in specific regions of a genome. Such ZFN-induced breaks can induce mutations in genes of interest through errorprone non-homologous end joining or lead to the modification of genes by homologous recombination in the presence of donor DNA or single-stranded oligonucleotides. Such targeted-genome editing approaches have been carried out across a variety of species, including fruit flies, nematodes, fish, rats, plants, and human cells. Genetic modifications derived from ZFN technology greatly facilitate the investigation of biological processes. In addition, ZFN technology is actively being studied as a means of advanced gene therapy to correct pathogenic genes.
anticancer agents. Several asymmetric, head-to-tail bibenzimidazole derivatives, such as Hoechst 33258 and Hoechst 33342, exhibited antitumor activity by binding to minor groove of DNA at three consecutive A:T base pairs, leading to the inhibition of Top 1 activity. In addition, the symmetric bibenzimidazole derivatives, containing two groups of benzimidazole linked in head-to-head fashion, have been reported that they bind DNA minor groove with extending the binding site to four A:T base pairs and exhibit antitumor activity. However, there is no report on the mechanism of action for their antitumor activity. Here, we showed that STK295900 exerted its activity by interfering with Top 1 and Top 2 activities. In support of this notion, STK295900 was recently reported as a potent antistaphylococcal agent by targeting DNA gyrase. The results from DNA relaxation assay suggested that STK295900 stabilizes the DNA-Top 1 cleavable complex, a characteristic of Top poisons, but it also inhibited Top 2 catalytic activity. Generally, Top poisons causes DNA strand break and consequently triggers G2 arrest via activation of ATM/ATR signaling pathway. These kinases phosphorylate and activate Chk1 and Chk2, which in turn phosphorylate and inactivate Cdc25C phosphatase resulting in blocking the activation of Cdk1 and transition into mitosis. Zinc finger nucleases are artificial restriction enzymes that are comprised of custom-designed zinc finger proteins and a nuclease domain derived from the FokI endonuclease. Zinc finger proteins can be designed to bind to specific sequences of DNA, allowing ZFNs to induce double- or single-strand breaks in specific regions of a genome. Such ZFN-induced breaks can induce mutations in genes of interest through errorprone non-homologous end joining or lead to the modification of genes by homologous recombination in the presence of donor DNA or single-stranded oligonucleotides. Such targeted-genome editing approaches have been carried out across a variety of species, including fruit flies, nematodes, fish, rats, plants, and human cells. Genetic modifications derived from ZFN technology greatly facilitate the investigation of biological processes. In addition, ZFN technology is actively being studied as a means of advanced gene therapy to correct pathogenic genes.
Author: targets inhibitor
Although the underlying mechanism of this effect is mainly due to the enhanced transduction of the virus
We found that ZFNs undergo proteasomal degradation and that MG132 increases ZFN levels, leading to enhanced genetic modifications by the ZFNs. Our protein stability study should lay the foundation for the advancement of ZFN technology; furthermore, the identification of MG132 as a small molecule that increases ZFN function is expected to facilitate the use of ZFNs. Targeted genetic modification using ZFNs can enable targeted gene insertion, correction, disruption, chromosomal rearrangement, 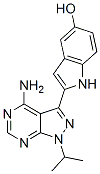 and regulatory region alteration. Gene editing using ZFNs is a promising technology as a powerful tool for studying biological processes and for the development of R428 advanced gene therapy to correct pathogenic genes. Here for the first time we investigated ZFN stability. Given that high levels of ZFN protein are associated with enhanced ZFN activity, our protein stability study should lay the foundation for further development of ZFN technology. Furthermore, ZFNs can be now delivered directly as protein and several doses of ZFN protein treatment are required to obtain sufficient ZFN activity, because the ZFNs are degraded within a few hours after treatment. Thus, the development of methods to maintain sufficient ZFN concentrations is important; our protein stability study should serve as a basis for this research. Even when a ZFN protein is continuously expressed by a DNA vector transfected into the target cells, inhibition of ZFN degradation increased the ZFN protein levels, leading to enhanced genetic modification. Porteus�� group has PB 203580 supply previously reported that short-term exposure to MG132 does not significantly increase the protein levels and activity of ZFNs that contain wild-type FokI nucleases. In contrast, we observed that MG132 treatment increased ZFN activity 2.4 or 2-fold. One possible reason for this discrepancy could be the different FokI nucleases employed in the two experiments: the ZFN used by Porteus�� group contained the wildtype FokI nuclease, whereas we used ZFNs with a modified FokI nuclease, which is improved from the wild type and is now predominantly used. This difference in the ZFN amino acid sequence might affect the rate of ZFN proteolysis. Another reason could be the difference in the MG132 concentration and duration of exposure: we treated cells with MG132 for 60 hours at 1, 2, and 5 mM, whereas Porteus�� group used 10 mM of MG132 for only 4 hours. We observed significantly decreased cell viability at 10 mM of MG132 when cells were treated for 60 hours. In addition, the application of MG132 to human embryonic stem cells caused cytotoxic effects even at very low dosage, which is compatible with the previous reports that showed similar cytotoxic effects of MG132 on hESCs. Here we showed that ZFN activity can be enhanced using a small molecule, MG132. To our knowledge, this is the first study reporting that a small molecule can regulate ZFN function. Identifying small molecules with this property is important given that ZFN technology is actively being studied as a tool for gene therapy and to analyze biological processes. Although MG132 is not a FDA-approved drug, other FDA-approved proteasomal inhibitors such as bortezomib might be used together with ZFNs to enhance the effect of gene therapy. Indeed, it has been recently reported that bortezomib can increase the effect of a ZFN-expressing adeno-associated virus.
and regulatory region alteration. Gene editing using ZFNs is a promising technology as a powerful tool for studying biological processes and for the development of R428 advanced gene therapy to correct pathogenic genes. Here for the first time we investigated ZFN stability. Given that high levels of ZFN protein are associated with enhanced ZFN activity, our protein stability study should lay the foundation for further development of ZFN technology. Furthermore, ZFNs can be now delivered directly as protein and several doses of ZFN protein treatment are required to obtain sufficient ZFN activity, because the ZFNs are degraded within a few hours after treatment. Thus, the development of methods to maintain sufficient ZFN concentrations is important; our protein stability study should serve as a basis for this research. Even when a ZFN protein is continuously expressed by a DNA vector transfected into the target cells, inhibition of ZFN degradation increased the ZFN protein levels, leading to enhanced genetic modification. Porteus�� group has PB 203580 supply previously reported that short-term exposure to MG132 does not significantly increase the protein levels and activity of ZFNs that contain wild-type FokI nucleases. In contrast, we observed that MG132 treatment increased ZFN activity 2.4 or 2-fold. One possible reason for this discrepancy could be the different FokI nucleases employed in the two experiments: the ZFN used by Porteus�� group contained the wildtype FokI nuclease, whereas we used ZFNs with a modified FokI nuclease, which is improved from the wild type and is now predominantly used. This difference in the ZFN amino acid sequence might affect the rate of ZFN proteolysis. Another reason could be the difference in the MG132 concentration and duration of exposure: we treated cells with MG132 for 60 hours at 1, 2, and 5 mM, whereas Porteus�� group used 10 mM of MG132 for only 4 hours. We observed significantly decreased cell viability at 10 mM of MG132 when cells were treated for 60 hours. In addition, the application of MG132 to human embryonic stem cells caused cytotoxic effects even at very low dosage, which is compatible with the previous reports that showed similar cytotoxic effects of MG132 on hESCs. Here we showed that ZFN activity can be enhanced using a small molecule, MG132. To our knowledge, this is the first study reporting that a small molecule can regulate ZFN function. Identifying small molecules with this property is important given that ZFN technology is actively being studied as a tool for gene therapy and to analyze biological processes. Although MG132 is not a FDA-approved drug, other FDA-approved proteasomal inhibitors such as bortezomib might be used together with ZFNs to enhance the effect of gene therapy. Indeed, it has been recently reported that bortezomib can increase the effect of a ZFN-expressing adeno-associated virus.
Comparing the respons fraction of ZFN-treated cells small molecules can be used in vitro to facilitate gene editing
In conclusion, we show that ZFN proteins have a relatively short half-life and that their turn-over is regulated by the UPP. Furthermore, treatment with the proteasome inhibitor MG132 blocked ZFN protein PF-4217903 cost degradation and extended its half-life, resulting in increased ZFN protein levels and enhanced genetic modification. Our protein stability study should lay the foundation for further development of ZFN technology. The identification of small molecules that increase ZFN protein levels will facilitate the application of ZFNs. To measure the level of functional miRNA in a manner that avoids detecting miRNA mimic trapped in non-functional locations, we immunoprecipitated UV cross-linked RISC from control and transfected cells and measured the amount of RISCassociated miR-200a by deep sequencing of the miRNA-sized RNA fraction in the immunoprecipitate. This revealed that the amount of RISC-associated miR-200a in the transfected cells was approximately equal to the level of other abundant miRNAs. This is proportionally much less than the level of miR200a measured by qPCR, indicating most of the transfected miRNA mimic is not bound to Argonaute and consequently is not functional. Similar results were obtained following transfection of a different miRNA, miR-200b. Thus, although qPCR is a valid technique to measure total miRNA amount, this can be very different from the amount of functional miRNA. Given the majority of miRNA mimic detected by qPCR did not represent the active Argonaute-bound population, we determined its sub-cellular localisation by transfecting a fluorescent siRNA and examining the transfected cells by fluorescence microscopy. The majority of the siRNA did not co-localise with Argonaute, which is consistent with earlier reports of transfected siRNA localising in large cytoplasmic aggregates that are distinct from the GW bodies that are known for their role in RNA silencing. Instead the vast majority of miRNA transfected with either HiPerfect,, RNAi-Max or Lipofectamine 2000 localised with or adjacent to lysosomes, matching earlier reports of lipid-based siRNA transfection. Therefore, the high level of transfected miRNA detected by qPCR is largely attributable to their retention within vesicles and subsequent amplification by qPCR following lysis. Hence, the use of qPCR to measure a miRNA after transient transfection can give the false impression that the miRNA is at massively nonphysiological level, whereas the amount of miRNA bound to Argonaute may indeed be appropriately physiological. On the other hand, it is conceivable that an inefficient ![]() transfection that results in just a small proportion of cells being transfected could appear to produce an adequate level of miRNA, if measured by qPCR. It is more appropriate to use an assay of miRNA function to verify the effectiveness of the transfection. Of additional interest to users of miRNA mimics for transient transfection, we were able to confirm from our sequencing of the Argonaute-bound pool of small RNAs, that while a miRNA mimic with unmodified passenger strand results in abundant incorporation of the passenger strand into RISC, raising the potential for extensive off-target effects, a mimic that is modified to limit the incorporation of the passenger strand into RISC does indeed achieve this. Although the merits of modified mimics have been previously recognised, published evidence for this is NVP-BKM120 limited to date and has been based largely on reporter assays.
transfection that results in just a small proportion of cells being transfected could appear to produce an adequate level of miRNA, if measured by qPCR. It is more appropriate to use an assay of miRNA function to verify the effectiveness of the transfection. Of additional interest to users of miRNA mimics for transient transfection, we were able to confirm from our sequencing of the Argonaute-bound pool of small RNAs, that while a miRNA mimic with unmodified passenger strand results in abundant incorporation of the passenger strand into RISC, raising the potential for extensive off-target effects, a mimic that is modified to limit the incorporation of the passenger strand into RISC does indeed achieve this. Although the merits of modified mimics have been previously recognised, published evidence for this is NVP-BKM120 limited to date and has been based largely on reporter assays.
We wished to check ZFN proteolysis with MG132 and determine the effects on mediated gene disruption
For example, the ZFN nuclease domain has been modified to improve ZFN activity and Dinaciclib CDK inhibitor specificity. Additionally, modifying the culture temperature caused a significant increase in ZFN activity. Furthermore, our group recently reported a simple method to enrich cells that contain ZFN-induced gene disruptions. Given that these simple methods to improve the ZFN function have facilitated the use of ZFNs, the identification of small molecules that increase ZFN function should likewise efficiently facilitate the application of ZFNs. However, such small molecules have yet to be identified. It has been observed that ZFN protein levels are directly correlated with ZFN function. Culturing the cells at low temperature increases ZFN function at least in part because ZFN protein levels increase. We also observed that cell populations that are enriched with gene-disrupted cells have high ZFN levels as compared to control cells. Recently, direct delivery of ZFN proteins has been shown to be safer associated with negligible offtarget effects. These ZFN proteins could penetrate the cells without any additional cell-penetrating peptide sequences and were able to transduce into several cell types including those that are hard to transfect. However, due to degradation of the delivered protein, it was necessary to treat the cells several times with the ZFN protein to obtain significant genetic modifications. Thus, we postulated that stabilizing the ZFN protein could enhance ZFN function. However, ZFN stability and the factors that affect it have yet to be investigated. Proteins are in a continual state of flux between synthesis and degradation in a cell. The ubiquitin proteasome pathway is one of the major cellular regulatory mechanisms involved in protein turnover and half-life. UPP plays a key role in eliminating intracellular proteins in eukaryotes, especially misfolded cellular proteins. During ubiquitination, a post-translational modification that targets proteins for degradation by the 26S proteasome, multiple ubiquitin molecules are covalently attached to targeted proteins. This process is catalyzed by a three step cascade mechanism, which involves a ubiquitin activating enzyme, a ubiquitin conjugating enzyme, and a ubiquitin ligase. E1 activates ubiquitin molecules by the formation of an ATP-dependent thiol ester bond between the C-terminus of ubiquitin and the active cysteine site of the E1 enzyme. Activated ubiquitin is transferred to the active cysteine site of the E2 enzyme. Ultimately, E3 catalyzes the transfer of ubiquitin molecules to a lysine residue, ultimately forming polyubiquitin chains on the protein that is  destined for degradation. Finally, ubiquitinated proteins are directed into the 20S core proteolytic KRX-0401 chamber in an ATP-dependent manner for 26S proteasomal degradation. Small chemical molecules, such as synthetic, cell-permeable peptide aldehydes that form covalent adducts with the 20S proteasome and inhibit its peptidase activities, have been developed. Synthetic proteasome inhibitors are peptide aldehydes which are broadly used as inhibitors for both Serine and Cysteine proteases. Several proteasome inhibitors that can enter the cells and block protein degradation pathway have been identified. Among them, the proteasome inhibitor MG132 is the most widely used commercial inhibitor for regulating the UPP. Because ZFN levels are directly proportional to ZFN activity.
destined for degradation. Finally, ubiquitinated proteins are directed into the 20S core proteolytic KRX-0401 chamber in an ATP-dependent manner for 26S proteasomal degradation. Small chemical molecules, such as synthetic, cell-permeable peptide aldehydes that form covalent adducts with the 20S proteasome and inhibit its peptidase activities, have been developed. Synthetic proteasome inhibitors are peptide aldehydes which are broadly used as inhibitors for both Serine and Cysteine proteases. Several proteasome inhibitors that can enter the cells and block protein degradation pathway have been identified. Among them, the proteasome inhibitor MG132 is the most widely used commercial inhibitor for regulating the UPP. Because ZFN levels are directly proportional to ZFN activity.
Sequence similarity and effect of the heart to protect against the toxic effects of doxorubicin
Downstream effectors of Akt and Erk converge to the mitochondria and initiate a protective response. There is additional evidence that coronary delivery of constitutively active form of Akt1 gene protects the heart against doxorubicininduced chronic heart failure by improving cardiac performance. We postulate that the increase in the pro-survival proteins observed in this study serves as an innate mechanism of the heart to protect against the damaging effects of doxorubicin. We also show increased p-Akt levels when treated with mdivi-1 alone and a BMS-354825 302962-49-8 further increase when treated with the combination of mdivi-1 and doxorubicin. A link between Akt pathway and the mitochondrial fusion and fission mechanism has been suggested previously. It is believed that increase in Akt phosphorylation promotes mitochondrial fusion, which is considered to lead to its cardioprotective effects. It has also been reported that compounds which offer cardio-protection such as insulin or anti-oxidants prevent ischaemia induced fragmentation and produces elongated mitochondria. It has also been speculated that the cytokine erythropoietin induces mitochondrial fusion by activating Akt. However, a downstream effector of Akt, protein kinase G, has been reported to phosphorylate and inhibit the pro-fission activity of Drp1. A recent study reported increase in the levels insulin stimulated Akt phosphorylation when also treated with mdivi-1. Further investigations are needed to establish whether mdivi-1 treatment causes a direct effect on Akt phosphorylation. We speculate that the huge increase observed in Akt phosphorylation when co-treated with doxorubicin and mdivi-1, is due to the dual effect of doxorubicin and a direct effect of mdivi-1 on Akt phosphorylation causing a further increase. Previously, we reported a significant increase in the levels of p-Akt following doxorubicin-treatment in conditions of ischaemia and reperfusion injury, which was partially blocked when coadministered with cyclosporin A as well as providing protection against the toxic effects of doxorubicin. Further studies are required to investigate the exact role of doxorubicin-induced toxicity and the protection there from with adjunct therapy on Akt phosphorylation. Studies have also reported a link between ROS generation, mitochondrial morphology and Erk 1/2 signalling in the regulation of insulin signalling pathway. Obesity induced ROS appeared to increase the levels of Erk 1/2 phosphorylation, which were reversed when treated with mdivi-1. A similar effect is also seen in our data showing that co-treatment with mdivi-1 reverses doxorubicin induced increase in Erk 1/2 levels. Furthermore, it has been reported that doxorubicin-induced involves Erk/p53 transduction pathway. Treatment of H9c2 and cardiac myocytes with doxorubicin caused an increase in the levels of p53 which were preceded by activation and nuclear translocation of Erk 1/2. They also showed that inhibition of Erk 1/2 with U-0126 prevented activation and nuclear translocation of both Erk 1/2 and p53 whilst inhibition of p53 with pifithrin-�� only prevented doxorubicin-induced changes in p53. In the current study we show an increase in the levels of p53 and Erk 1/2 following treatment with doxorubicin. Interestingly, co-administration of doxorubicin with mdivi-1, which prevented detrimental effects of doxorubicin in the  Langendorff and oxidative stress model and reduced the levels of p-Drp1, also prevented the increase in the levels of both p53 and Erk 1/2. Further studies are required to identify the specific role of Erk 1/2 and Akt activation using their specific inhibitors and the CP-690550 intracellular signalling pathway associate with the protective effect mdivi-1. Furthermore, we show that co-treatment with mdivi-1 does not interfere with the anti-cancer properties of doxorubicin as assessed by MTT assay using HL60 cells. It is imperative to assess the effects of adjunct therapies, aiming to reduce the cardiotoxic effect, on the anti-tumour effects. Many cardioprotective strategies fail to demonstrate beneficial effects in clinical or in vivo settings as they interfere or reduce with the anti-cancer effects and thereby reduce the clinical utility. Collectively, our data show that co-treatment with the mitochondrial fission inhibitor mdivi-1 can ameliorate the cardiotoxic effects of doxorubicin without affecting its anticancer properties. These finding warrant further investigations in the relevant animal models of cancer. The p21-activated kinase family comprises six sterile-20 group serine/threonine kinases.
Langendorff and oxidative stress model and reduced the levels of p-Drp1, also prevented the increase in the levels of both p53 and Erk 1/2. Further studies are required to identify the specific role of Erk 1/2 and Akt activation using their specific inhibitors and the CP-690550 intracellular signalling pathway associate with the protective effect mdivi-1. Furthermore, we show that co-treatment with mdivi-1 does not interfere with the anti-cancer properties of doxorubicin as assessed by MTT assay using HL60 cells. It is imperative to assess the effects of adjunct therapies, aiming to reduce the cardiotoxic effect, on the anti-tumour effects. Many cardioprotective strategies fail to demonstrate beneficial effects in clinical or in vivo settings as they interfere or reduce with the anti-cancer effects and thereby reduce the clinical utility. Collectively, our data show that co-treatment with the mitochondrial fission inhibitor mdivi-1 can ameliorate the cardiotoxic effects of doxorubicin without affecting its anticancer properties. These finding warrant further investigations in the relevant animal models of cancer. The p21-activated kinase family comprises six sterile-20 group serine/threonine kinases.