We and other groups have previously shown that OGlcNAcylation regulates phosphorylation of tau in a reciprocal manner overall. Our observations of decreased tau phosphorylation at Thr181, Thr212, Ser214, Ser262/Ser356, Ser404 and Ser409 are consistent with previous studies, further supporting the reciprocal relationship between O-GlcNAcylation and phosphorylation of tau at these sites. To understand the underlying mechanism by which FDA-approved Compound Library thiamet-G increased tau phosphorylation at some other phosphorylation sites in the mouse brain, we investigated the effects of thiamet-G injection on the major tau kinases. We found that thiamet-G at the dose injected into the brain completely blocked Ser9 phosphorylation of GSK3b, indicating a marked activation of this major tau kinase. Our previous studies have demonstrated that tau phosphorylation at Ser199, Ser202 and Ser396 are mainly regulated by GSK-3b. Taken together, we conclude that the elevation of tau phosphorylation at Ser199, Ser202, Ser396 and Ser422 results from the combinational effects of the increase in tau O-GlcNAcylation and the activation of GSK-3b activity, and that GSK-3b activation predominated and led to the net increase in tau phosphorylation at these sites in the mouse brain. Our results showed that GSK-3b was not activated in cultured neuronal cells treated with thiametG, consistent with the absence of any increase in tau phosphorylation at these phosphorylation sites. In a previous study, when thiamet-G was administered to rats orally for 24 hrs, tau phosphorylation at these sites was not found to be increased. Whether the discrepancy between this previous study and the present study is due to different routes of drug administration, the attainment of different doses within the brain, or the use of different speciesis currently unknown. It is possible that there is either a dose-dependent effect of MK-2206 1032349-77-1 thiametG on 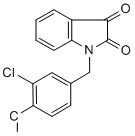 GSK-3b�Cstimulation or an off target effect of thiamet-G when used at high doses directly in the brain. Indeed, it is likely that the icv injection of this study led to a much higher thiamet-G concentration in the central nervous system than that from oral dosing. Unfortunately, GSK-3b modification and activity in the brain was not examined in the previous study, so direct comparisons are not possible. Our results indicated marked differences in the effects of thiamet-G on tau phosphorylation between the mouse brains and the cultured neurons. Further experiments indicated that thiametG�Cinduced increase of tau phosphorylation at several sites resulted from activation of GSK-3b, a major tau kinase, but this activation did not occur in cultured cells. Different regulations of tau phosphorylation by extracellular signaling between the brains and the cultured neurons might also contribute to the different results we observed. A previous study has demonstrated that tau phosphorylation is regulated by FGF-2 through GSK-3b. Our observations of GSK-3b activation in the brains but not in the cultured cells are consistent with this speculation. The mice we used for this study expressed the wild-type human tau isoform. Although we cannot absolutely rule out the possibility that the different results between the mouse brains and the cell cultures might also result from different tau species, this possibility is very small, because our observations that thiamet-G�Cinduced GSK-3b activation in the brains, but not in the cultured cells, can explain the different results in these two systems nicely. Our studies on the upstream regulating kinases of GSK-3b suggest that thiamet-G led to marked GSK-3b activation.
GSK-3b�Cstimulation or an off target effect of thiamet-G when used at high doses directly in the brain. Indeed, it is likely that the icv injection of this study led to a much higher thiamet-G concentration in the central nervous system than that from oral dosing. Unfortunately, GSK-3b modification and activity in the brain was not examined in the previous study, so direct comparisons are not possible. Our results indicated marked differences in the effects of thiamet-G on tau phosphorylation between the mouse brains and the cultured neurons. Further experiments indicated that thiametG�Cinduced increase of tau phosphorylation at several sites resulted from activation of GSK-3b, a major tau kinase, but this activation did not occur in cultured cells. Different regulations of tau phosphorylation by extracellular signaling between the brains and the cultured neurons might also contribute to the different results we observed. A previous study has demonstrated that tau phosphorylation is regulated by FGF-2 through GSK-3b. Our observations of GSK-3b activation in the brains but not in the cultured cells are consistent with this speculation. The mice we used for this study expressed the wild-type human tau isoform. Although we cannot absolutely rule out the possibility that the different results between the mouse brains and the cell cultures might also result from different tau species, this possibility is very small, because our observations that thiamet-G�Cinduced GSK-3b activation in the brains, but not in the cultured cells, can explain the different results in these two systems nicely. Our studies on the upstream regulating kinases of GSK-3b suggest that thiamet-G led to marked GSK-3b activation.