Predicted to phosphorylate Bcl2-associated athanogene 3, Maf1 and peroxisome biogenesis factor 1. In recent years, several groups have performed comprehensive tissue phosphoproteome analyses. The animal tissue most often used for phosphoproteome analysis has been liver. The first such study, performed by Jin et al., utilized iron IMAC for the enrichment of phosphorylated peptides and conventional linear ion-trap mass spectrometry for the phosphopeptide analysis. These investigators identified 26 nonredundant phosphorylation sites. A subsequent study utilized high capacity iron IMAC and a higher mass accuracy MS Q-TOF instrument to identify 339 non-redundant phosphorylation sites from over 200 proteins. The analyses performed by Villen et al. in 2007 was a breakthrough study. These investigators identified 5,635 nonredundant phosphorylation sites from 2,149 proteins, signifying the first in-depth global analysis of phosphopeptides from liver. Notably, these investigators used more selective iron IMAC beads and MS instruments of higher mass accuracy as compared to previous studies. However, it is likely that the SCX pre-fractionation of tryptic peptides was a critical factor accounting for the increase in phosphopeptide identification. Lack of reproducibility has been an issue with large scale MSbased phosphoproteomic profiling. Moser and White tested the reproducibility of their methodology by performing three replicate analyses of the same rat liver homogenate. They observed that 56�C63% of the peptides from each analysis were observed in all three analyses. Another issue is method of analysis. Alcolea et al. found that analyzing the same phosphopeptide enriched murine NIH/3T3 fibroblast lysate by two different LC-MS/MS Gomisin-D platforms based on Q-TOF and LTQ-Orbitrap mass spectrometers led to identification 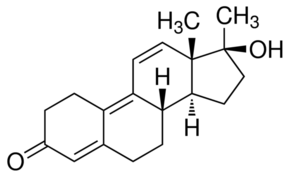 of partially overlapping, but also distinct, phosphoproteome profiles. The QTOF based platform resulted in 1,485 non-redundant phosphopeptide identifications, whereas the LTQ-Orbitrap based platform identified 4,308 non-redundant phosphopeptides. Only 1,077 of the total population of phosphopeptides were detected by both platforms. Analyzing duplicate samples by LC-MS/MS on the LTQ-Orbitrap platform showed that,70% of the identified phosphopeptides were identical. In a study comparing the peptides identified using two workflows, TiO2-SCX and SCX-TiO2, the overlap was 58 and 51% for the two methods, respectively. A similar overlap of 60% was observed, when they performed replicate LC-MS/MS analyses of the same TiO2-SCX sample by an LTQ-Orbitrap. These previous studies profiling the liver phosphoproteome have not been aimed at characterizing signaling events in a physiological context. Accomplishing this required LOUREIRIN-B improved sensitivity and accuracy, and a demonstration of reproducibility. We found that peptide abundance affects reproducibility, but that reproducibility could be enhanced by the performance of technical replicates. There are several indications that our methods were sufficient to detect mTORC1-mediated protein phosphorylation. These included the identification of known mTORC1 targets, the significant enrichment for mTOR signaling pathway constituents as indicated by pathway analysis, and the kinase prediction results. In their seminal studies, Hunter and Sefton reported the relative abundances of pSer, pThr, and pTyr to be 90%, 10%, and 0.05%,respectively. The order of magnitude higher tyrosine phosphorylation frequency in our results may be a function of the tendency for pTyr-containing peptides to yield better quality MS/MS spectra with collision-induced dissociation fragmentation.
of partially overlapping, but also distinct, phosphoproteome profiles. The QTOF based platform resulted in 1,485 non-redundant phosphopeptide identifications, whereas the LTQ-Orbitrap based platform identified 4,308 non-redundant phosphopeptides. Only 1,077 of the total population of phosphopeptides were detected by both platforms. Analyzing duplicate samples by LC-MS/MS on the LTQ-Orbitrap platform showed that,70% of the identified phosphopeptides were identical. In a study comparing the peptides identified using two workflows, TiO2-SCX and SCX-TiO2, the overlap was 58 and 51% for the two methods, respectively. A similar overlap of 60% was observed, when they performed replicate LC-MS/MS analyses of the same TiO2-SCX sample by an LTQ-Orbitrap. These previous studies profiling the liver phosphoproteome have not been aimed at characterizing signaling events in a physiological context. Accomplishing this required LOUREIRIN-B improved sensitivity and accuracy, and a demonstration of reproducibility. We found that peptide abundance affects reproducibility, but that reproducibility could be enhanced by the performance of technical replicates. There are several indications that our methods were sufficient to detect mTORC1-mediated protein phosphorylation. These included the identification of known mTORC1 targets, the significant enrichment for mTOR signaling pathway constituents as indicated by pathway analysis, and the kinase prediction results. In their seminal studies, Hunter and Sefton reported the relative abundances of pSer, pThr, and pTyr to be 90%, 10%, and 0.05%,respectively. The order of magnitude higher tyrosine phosphorylation frequency in our results may be a function of the tendency for pTyr-containing peptides to yield better quality MS/MS spectra with collision-induced dissociation fragmentation.